







With advancing age, most regenerating organs suffer a cumulative decline in functional capacity that is associated with an increase in age-associated diseases such as cancer. These phenomena are thought to be driven by defects at the level of adult stem cells, whose progressive attrition with age is likely to precipitate tissue dysfunction. Adult stem cells are also the likely cell of origin for many cancers. There are multiple lines of evidence implicating the accumulation of DNA damage within adult stem cells as causative for these age-associated phenotypes. Firstly, many accelerated ageing disorders (progeroid syndromes), such as Hutchinson-Gilford progeria, Werner syndrome, Bloom’s syndrome, xeroderma pigmentosum, Cockayne syndrome, dyskeratosis congenita and Fanconi anemia, are caused by defects leading to elevated levels of DNA damage and/or in the compromised repair of DNA damage. Secondly, DNA damage has been shown to provoke cell fate decisions in adult stem cells that would be consistent with depletion with age. These include apoptosis, senescence, differentiation and malignant transformation. Thirdly, DNA mutations have been shown to accumulate in adult stem cells in an age dependent manner, with some estimates predicting that human HSCs acquire around 14 mutations per year per stem cell, which equates to an average mutation burden of around 700 mutant alleles in each HSC by the time an individual reaches the age of 50. Finally, pre-malignant oncogenic mutations have been found to exist in the normal stem cell pool of aged individuals. These mutations impact on HSC behavior, and can drive the non-malignant clonal outgrowth of individual HSC clones, which is associated with an increased risk of developing hematologic malignancy.
Although DNA damage within stem cells is well accepted as an important mechanistic cause of tissue ageing, the main physiologic source of DNA damage within the stem cell compartment is not clear. A more thorough understanding of the environmental factors that are responsible for DNA damage in these rare but important cell populations would potentially allow better assessment of the risk of a individual developing either cancer or other age related pathologies. One could also imagine that this knowledge would also facilitate the development of therapeutic strategies to prevent or delay this process.
Environmental stress is a biologically relevant mediator of DNA damage in the HSC compartment
|
|
A rude awakening for stem cells: Various forms of environmental stress, such as infection, inflammation or blood loss, can force HSCs to leave their so-called “dormant” state and enter into active cell cycle in order to replenish mature blood cells. This period of forced exit from dormancy corresponds to conditions of high replication stress within HSCs, leading to DNA damage and HSC depletion. |

A phenomenon known as replication stress is one potential source of DNA damage that is broadly applicable to most cell types across all individuals. Replication stress can be defined as any setting where the DNA replication fork stalls, increasing the likelihood of DNA strand breaks if the fork subsequently collapses. A number of biological phenomena can contribute to elevated levels of replication stress, including nucleotide pool imbalances, the presence of DNA adducts, hybrid DNA-RNA structures called R-loops and the presence of highly repetitive DNA sequences or complex secondary structure, all of which can inhibit the progression of the replication fork along DNA during S phase. However, it goes without saying that replication stress is only relevant in the context of actively proliferating cells and this poses a problem when we come to consider HSCs, which are characterized by very low levels of cell cycle activity under homeostatic conditions. In fact, the functional potency of HSCs has been shown to positively correlate with this status of long-term quiescence, also known as dormancy. We therefore hypothesized that HSC dormancy might be a route via which the genomic and associated functional integrity of HSCs is preserved during ageing, by acting to limit replication stress. This dormant state can however be broken under certain physiologic conditions, such as during infection or following blood loss, where HSCs are forced into cycle in order to regenerate the blood system. In a recent publication in the journal Nature1, we have shown that the exposure of mice to various forms of environmental stress can lead to DNA damage in HSCs as a result of their exit from their homeostatic dormant status. Importantly, the biological relevance of this finding to ageing was made clear when we observed that chronic stress led to a depletion in the number of functional HSCs, resulting in a phenotype akin to accelerated ageing of the blood system.
The Fanconi anemia DNA repair pathway is an important route via which stress-induced DNA damage is resolved.
|
|
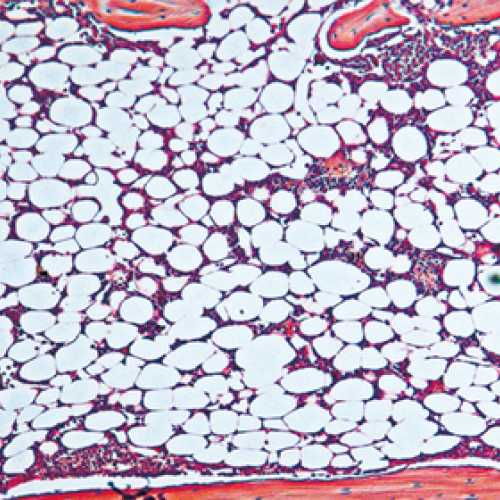
Stress hematopoiesis leads to bone marrow failure in DNA-repair defective Fanconi anemia knockout mice: Fanconi anemia knockout mice subject to chronic inflammatory stress (bottom panel) demonstrate severe bone marrow aplasia compared to their wild type counterparts subject to the same treatmenregimen (top panel). The image shows H&E stained bone marrow sections, with different types of blood cells being clearly very abundant in the wild type marrow, while in the FA knockout marrow, the blood cells have been depleted to be replaced by adipocytes. |
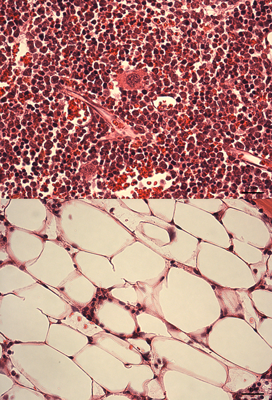
Fanconi anemia (FA) is the most common inherited bone marrow failure syndrome, resulting from inactivating mutations in any of up to 18 genes, which together form part of an epistatic signaling pathway. This pathway is involved in resolving certain forms of DNA damage related to stalled replication forks. Most FA patients develop early onset bone marrow failure that is thought to be driven by accelerated depletion of the HSC pool, leading to the disease being considered as a type of segmental progeria syndrome (reviewed in 2). In line with this accelerated ageing phenotype, FA patients also have an extremely high risk of developing leukemias and solid tumors. Mouse knockout models of FA have exactly the same DNA repair defect as that observed in human patients, but they never develop bone marrow failure, even at extreme old age3. This is likely due to the FA mice not encountering the source of DNA damage that is responsible for HSC attrition in FA patients during ageing. We hypothesized that HSC dormancy may protect FA knockout murine HSCs from DNA damage and that environmental stress may ultimately be the missing source of DNA damage in experimental mice. In line with this concept, we could show that HSCs from FA knockout mice demonstrated elevated levels of DNA damage in vivo in response to various forms of physiologic stress, suggesting that the FA pathway is an important route via which this damage is resolved. Importantly, if we repeated the chronic stress treatment that resulted in accelerated ageing within the hematopoietic system of wild type mice, this led to an almost complete exhaustion of the HSC pool in FA mice and precipitated a highly penetrant bone marrow failure phenotype, highly reminiscent of that observed in patients1. We believe that this new model will not only allow us to better understand the etiology of this devastating disease, but will also permit us to more broadly gain new insight into the process of ageing and malignant transformation across the general population.
Key Publications on DNA damage in HSCs and Fanconi anemia
1. Walter D., Lier A, Geiselhart A., Thalheimer F.B., Huntscha S., Sobotta M.C., Moehrle B., Brocks D., Bayindir I., Kaschutnig P., Müdder K., Klein C., Jauch A., Schroeder T., Geiger H., Dick T.P., Holland-Letz T., Schmezer P., Lane S.W., Rieger M.A., Essers M.A.G., Williams D.A.W., Trumpp A.and Milsom M.D. (2015). Exit from dormancy provokes DNA damage-induced attrition in haematopoietic stem cells. Nature, 520(7548): pp549-552).
2. Geiselhart A., Walter D., Lier A. and Milsom M.D. (2012). Disrupted signalling through the Fanconi anemia pathway leads to dysfunctional hematopoietic stem cell biology: underlying mechanisms and potential therapeutic strategies. Anemia, 2012: 265790.
3. Kaschutnig P., Bogeska R., Walter D., Lier A., Huntscha S. and Milsom M.D. (2015). The Fanconi anemia pathway is required for efficient repair of stress-induced DNA damage in hematopoietic stem cells. Cell Cycle, 14(17): pp2734-2742.
4. §Müller L.U.W., §Milsom M.D., Harris C.E., Vyas R., Brumme K.M., Parmar K., Moreau L.A., Schambach A., Park I.H., London W.B., Strait K., D’Andrea A., Daley G.Q. and Williams D.A. (2012). Overcoming reprogramming resistance of Fanconi anemia cells. Blood. 119 (23): pp5449-57. §Equal contribution.
5. Milsom M.D., Schiedlmeier B., Bailey J., Kim M.O., Li D., Jansen M., Ali M.A., Kirby M., Baum C., Fairbairn L.J. and Williams D.A. (2009). Ectopic HOXB4 overcomes the inhibitory effects of tumor necrosis factor-α on Fanconi anemia hematopoietic stem and progenitor cells. Blood, 113 (21): pp5111-20.
6. Milsom M.D., Lee A.W., Zheng Y. and Cancelas J.A. (2009). Fanca-/- hematopoietic stem cells demonstrate a mobilization defect which can be overcome by administration of the Rac inhibitor NSC23766. Haematologica, 94 (7): pp1011-15.
7. Milsom M.D., Jerabek-Willemsen M., Harris C.E., Schambach A., Broun E., Bailey J., Jansen M., Schleimer D., Nattamai K., Wilhelm J., Watson A., Geiger H., Margison G.P., Moritz T., Baum C., Thomale J., and Williams D.A. (2008). Reciprocal relationship between O6-methylguanine-DNA methyltransferase P140K expression level and chemoprotection of hematopoietic stem cells. Cancer Res., 68 (15): pp6171-80.
8. Müller L.U., Milsom M.D., Kim M.O., Schambach A., Schuesler T. and Williams D.A. (2008). Rapid lentiviral transduction preserves engraftment potential of Fanca-/- hematopoietic stem cells. Mol. Therapy, 16 (6): pp1154-60.
9. Southgate T.D., Sheard V., Milsom M.D., Ward T.H., Mairs R.J., Boyd M. and Fairbairn L.J. (2006). Radioprotective gene therapy through retroviral expression of manganese superoxide dismutase. J. Gene Med., 8 (5): pp557-65.
10. Milsom M.D., WoolfordL.B., MargisonG.P., HumphriesR.K. and Fairbairn L.J. (2004).Enhanced In Vivo Selection of Bone Marrow Cells by Retroviral-Mediated Co-expression of Mutant O6-Methylguanine-DNA-Methytransferase and HOXB4. Mol. Therapy, 10 (5): pp862-73.