







|
|
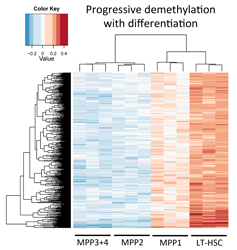
A number of differentially methylated regions (DMRs) of the genome can be identified between long-term Genome-wide analysis of these DMRs provides a picture of how these loci either progressively gain DNA methylation |
In addition to the cumulative acquisition of DNA mutations with increasing age, changes in chromatin structure can also enforce stable changes in gene expression programs, that can in turn lead to altered cell biology and disease. Indeed, abnormal functionality in enzymes responsible for regulating the levels of DNA methylation and/or histone methylation/acetylation have been identified as causative for cellular transformation and, in the setting of the hematopoietic system, an age-associated emergence of non-malignant dominant HSC clones. Clearly, progressive epigenetic changes within both pluripotent stem cells and adult stem cells are an important regulatory step, which enforces programs of cellular differentiation. Therefore the analysis of the HSC and hematopoietic progenitor epigenome holds the potential to give a new level of mechanistic insight into the process of normal hematopoietic differentiation as well as into abnormal hematopoiesis across a range of pathologic settings. However, such analyses are severely restricted by the low abundance of HSC and progenitor populations within the bone marrow, with approximately 1 in 100,000 murine bone marrow cells being an HSC.
|
|
Detailed locus specific analysis of DMRs can yield information relating to differentiation stage specific changes in DNA methylation levels thatmay relate to finely regulated gene expression changes. |
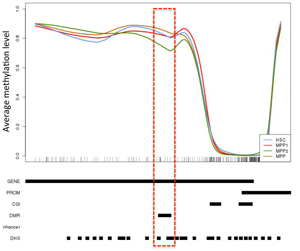
We have established an extremely productive collaboration with the groups of Prof. Christoph Plass (Division of Epigenomics and Cancer Risk Factors, DKFZ) and Dr. Daniel Lipka (Junior Research Group Regulation of Cellular Differentiation, DKFZ) in order to interrogate epigenetic changes in HSCs and hematopoietic progenitors, using new technological approaches that allow analysis of the epigenome using the limited amounts of starting material that can be harvested from HSCs and their immediate progeny. Together, we have recently been successful in generating genome wide DNA methylation data for highly purified murine HSCs and several MPP populations using a tagmentation-based whole genome bisulphite sequencing approach which allows increased efficiency of sequencing library preparation compared to conventional bisulphite sequencing. As part of a multi-group consortium, this data was combined with gene expression and proteome data from the same cell populations and published as a comprehensive resource article in the journal Cell Stem Cell1 in 2014. Along with a follow up publication in Cell Cycle2, this work provides a unique insight into the differential regulation of gene expression across several highly related cell populations that together form the apex of the murine hematopoietic system.
Key Publications on Epigenetic Regulation of Normal and Diseased HSCs and Leukemogenesis.
1. Cabezas-Wallscheid N., Klimmeck D., Hansson J., Lipka D.B., Reyes A., Wang Q., Weichenhan D., Lier A., von Paleske L., Renders S., Wünsche P., Brocks D., Gu L., Herrmann C., Haas S., Essers M., Brors B., Eils R., §Huber W., §Milsom M.D., §Plass C., §Krijgsveld J. and §Trumpp A. (2014). Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome and DNA methylome analysis. Cell Stem Cell, 15(4): pp507-522. §Shared senior authorship.
2. Lipka D.B., Wang Q., Cabezas-Wallscheid N, Klimmeck D, Weichenhan D., Herrmann C, Lier A., Brocks D., Assenov Y., von Paleske L., Renders S., Wünsche P., Zeisberger P., Gu L., Haas S., Essers M.A.G, Brors B, Eils R., §Trumpp A, §Milsom M.D., and §Plass C. (2014). Identification of DNA methylation changes at cis-regulatory elements during early steps of HSC differentiation using tagmentation-based whole genome bisulphite sequencing. Cell Cycle, 13(22): pp3476-3487. §Shared senior authorship.
3. Sonnet M., Claus R., Becker N., Zucknick M., Peterson J., Lipka D.B., Oakes C.C., Andrulis M., Lier A., Milsom M.D., Witte T., Gu L., Kim-Wanner S.-Z., Schirmacher P., Wulfert M., Gattermann N., Lübbert M., Rosenbauer F., Rehli M., Bullinger L., Weichenhan D. and Plass C. (2014). Early aberrant DNA methylation events in a mouse model of acute myeloid leukemia. Genome Medicine, 6 (4): 34
4. Placke T., Faber K., Nonami A., Putwain S., Salih H., Heidel F., Krämer A., Root D., Barbie D., Armstrong S., Hahn W., Huntly B., Sykes S., Milsom M.D., Scholl C. and Fröhling S. (2014). Requirement for CDK6 in MLL-rearranged acute myeloid leukemia. Blood, 124 (1): pp13-23